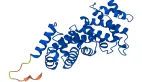
AlphaFold 2再發蛋白質結構預測結果,解鎖98.5%人類蛋白質組
上周在科學界發生了十分有戲劇性的一幕:兩大科學頂刊Science和nature同時發文,兩支研究團隊都表示可以通過AI模型預測蛋白質和一些分子復合物的精確3D原子結構。
來自華盛頓大學的科研團隊在Science上公布了RoseTTAFold,宣稱這個模型的性能與AlphaFold 2相當。
并且其端到端版本在RTX2080上生成少于400個殘基的蛋白質的骨干坐標僅需10分鐘。

RoseTTAFold蛋白質預測工具的論文
這顯然讓谷歌DeepMind團隊有些措手不及,匆匆忙忙選擇在nature上開源了AlphaFold 2的技術細節。
不過今天故事似乎要反轉:DeepMind團隊在nature上再接再厲,公布了他們已經使用AlphaFold工具預測的來自人類和20種其他生物共350000種蛋白質的結構,例如大腸桿菌、酵母菌和果蠅等等。

論文鏈接:https://www.nature.com/articles/s41586-021-03828-1
目前他們已經復現了44%的人類蛋白質結構,覆蓋了人類基因組編碼的所有氨基酸結構位置的近60%。
不僅如此,這個團隊將在未來的幾個月里,通過計算預測目前UniRef目錄中所有科學界已知的,經過測序的1億個蛋白質分子結構。
而AlphaFold與EMBL-EBI(歐洲分子生物學實驗室)合作推出了蛋白質結構數據庫(鏈接:
https://alphafold.ebi.ac.uk/),科研人員只要輸入蛋白質名稱或編號,就可以迅速獲得蛋白質的3D結構圖像。
模型好不好,還得看療效。看來AlphaFold這一次靠「鈔能力」贏了。
AlphaFold新進展
昨日,DeepMind在nature上刊登的文章,描述了AlphaFold對人類蛋白質組的結構預測,覆蓋了98.5%的所有人類蛋白質組,還對20種其它生物蛋白質的結構進行了預測。
這20種其它生物包括大腸桿菌、酵母菌、果蠅等,它們都是生物研究的重點。
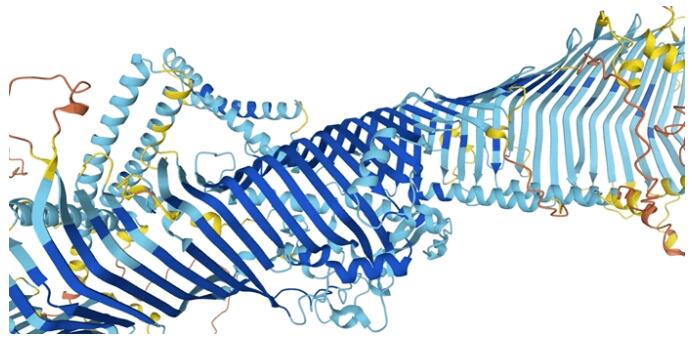
果蠅蛋白質3D結構示意圖
在論文中,作者表示,AlphaFold能夠對人類蛋白質組35.7%氨基酸結構位置進行預測,且有很高的置信度,是實驗方法覆蓋的結構數量的兩倍。
在單個蛋白質水平上,AlphaFold能夠對43.8%的蛋白中,至少四分之三的氨基酸序列給出了可信預測。

在單個蛋白質的介紹中,數據庫列出了蛋白質的基本信息,包括蛋白質名稱、基因、來源生物、生物功能等。

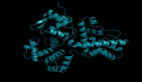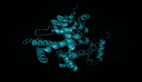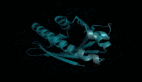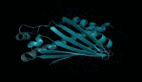
以T細胞免疫調節蛋白同源物為例,這個蛋白質可以通過免疫調節保護寄生蟲免受宿主免疫系統的攻擊。
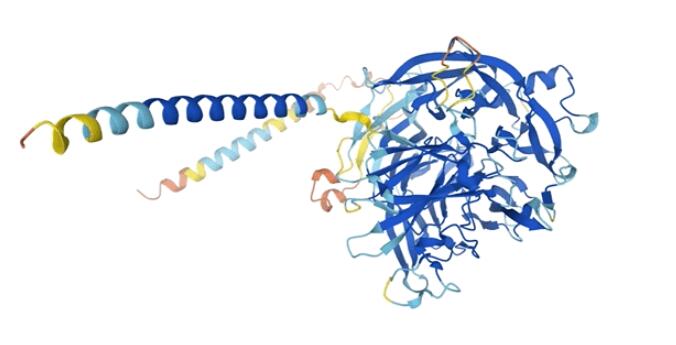
惡性瘧原蟲T細胞免疫調節蛋白同源物結構圖
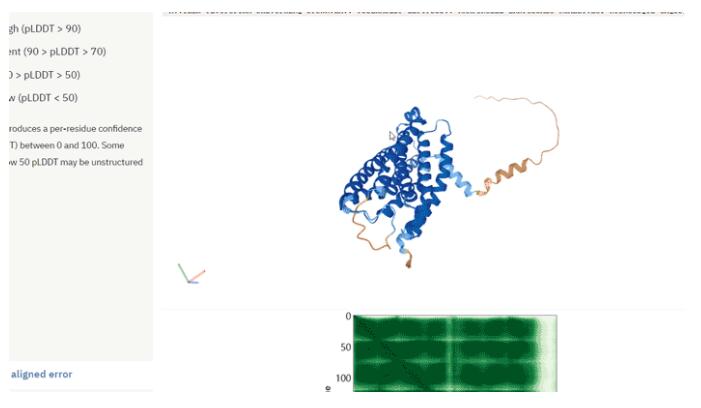
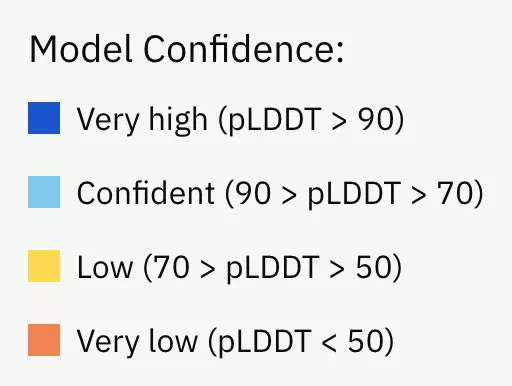
如上圖所示,DeepMind在數據庫公布的蛋白質結構預測模型中,不同的顏色表示不同的預測可信度。
殘基置信度分數(pLDDT)范圍在0-100,數值大于90指信度非常高,表示為藍色;數值為70-90表示為淺藍色;數值為50-70表示為黃色;橙色部分表示信度非常低,數值小于50,有可能是孤立的非結構化區域。
除了公布蛋白質基本信息及模型結構外,網站還公布了蛋白質結構的預測對齊誤差,后續還會推出蛋白質序列特征功能。
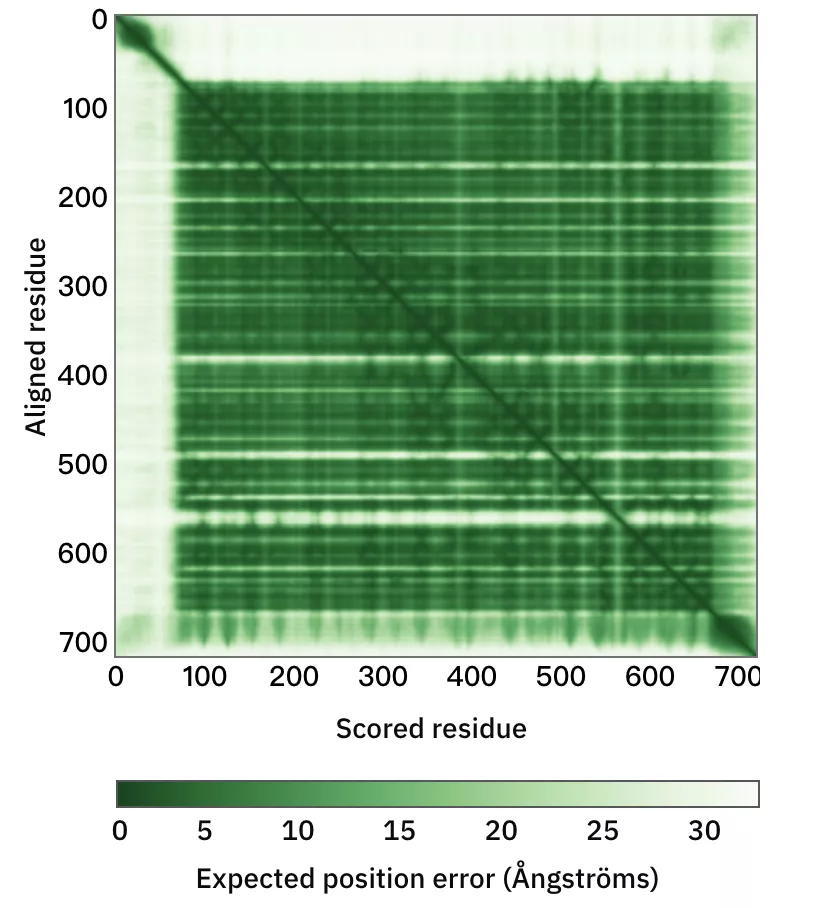
蛋白質結構模型的預測對齊誤差
AlphaFold 2預測出了約20000種人類基因組蛋白質,加上20種其它生物的蛋白質組的結構預測,總計公布了超過350000種蛋白質結構預測模型。
「AlphaFold確定了覆蓋幾乎整個人類蛋白質組(98.5%的所有人類蛋白,即Uniprot里所有2700aa以下的蛋白)的蛋白質的結構。」
DeepMind表示,接下來的幾個月將計劃擴大蛋白質模型列表,以涵蓋所有編目的蛋白質,共計大約1億個分子。
AlphaFold為近44%的人類蛋白質預測了結構,涵蓋了人類基因組中60%的氨基酸結構位置預測。
AlphaFold確定了許多其他人類蛋白質是「無序的」,也就是說,蛋白質的形狀不采用單一結構。
當這種無序的蛋白質與另一蛋白質結合時,最終可能會確定出一種結構。
當然,這些蛋白質也可能會有多種結構。
蛋白質結構預測:50年的難題
蛋白質的 3D 結構在很大程度上決定了它的功能,通過研究蛋白質的3D結構,生物學家不僅可以更加快速地研發出針對各類疾病的藥物,甚至能夠揭開生命之謎。
弄清楚蛋白質折疊成什么形狀被稱為“蛋白質折疊問題”,在過去的50年里一直是生物學的一個巨大挑戰。
通過結構測定是一個有效但非常費力的方法,它比基因組測定難度大得多。
按照傳統的實驗步驟,從基因序列到相應的蛋白質結構測定之間還要經過基因表達、蛋白質的提取和純化、結晶、X射線衍射分析等步驟。由于蛋白質結構和性質的多樣性,這些步驟大多沒有固定的規律可循。
并且試驗需要反復進行多次,每個結構的測定可能都需要數年的工作,并且動用價值數百萬元的設備。
在1972年諾貝爾化學獎的獲獎感言中,克里斯蒂安·安芬森( Christian Anfinsen ) 提出了一個著名假設:理論上,蛋白質的氨基酸序列應該完全決定其結構。

但是從理論上分析,蛋白質在形成之前,可以折疊出天文數字的3D結構。
1969年,賽勒斯·萊文塔爾 (Cyrus Levinthal) 指出,通過蠻力計算列舉出典型蛋白質的所有可能構型所需的時間比已知宇宙的年齡還要長。
他預測,一個典型的蛋白質可能有10的300次方種折疊形式。
如今DeepMind的團隊用AI解開了這道世紀難題。
經過訓練的深度神經網絡可以根據蛋白質的基因序列預測蛋白質的特性。主要判斷依據是氨基酸對之間的距離和連接這些氨基酸的化學鍵之間的角度。
通過已知信息,可以推斷出蛋白質折疊之后的角度和距離信息,從而推斷出整個蛋白質的結構。
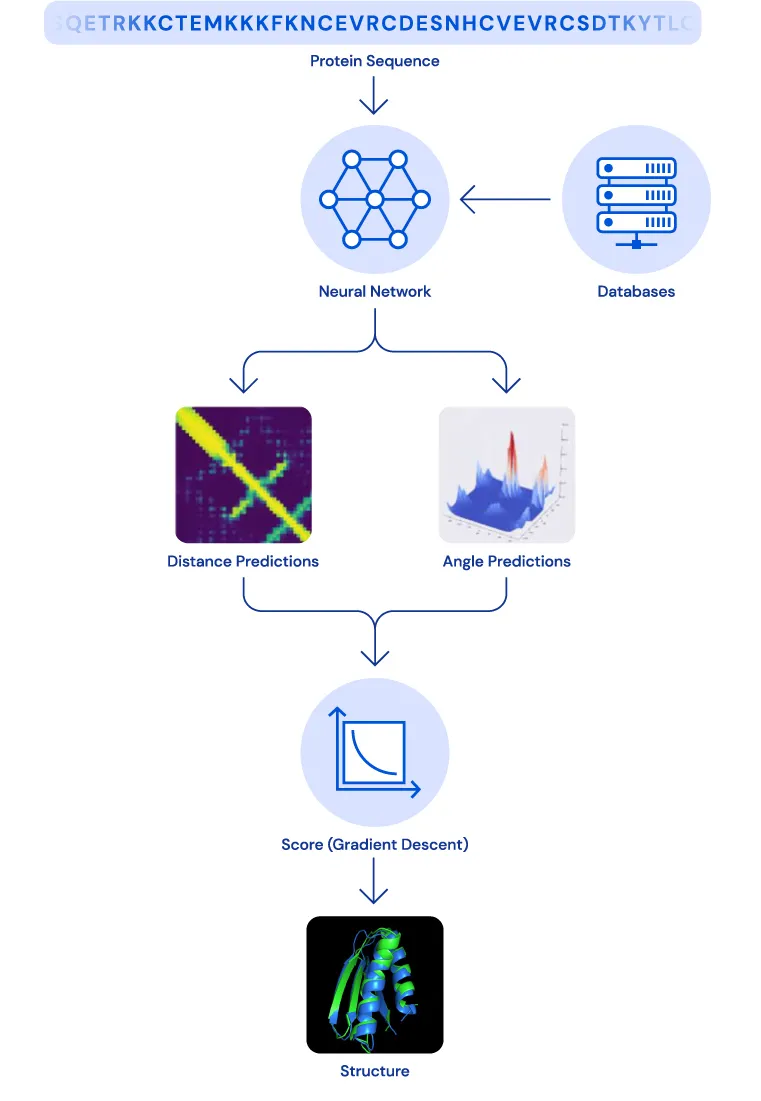
AlphaFold數據庫的建立,把生物科學家們從研究蛋白質時最費時費力的結構測定工作中解脫了出來,從而可以專心轉向研究的核心部分。
「我們原來花費數個月甚至數年時間才完成的事情,AlphaFold在一個周末就能完成」,樸茨茅斯大學結構生物學教授兼酶創新中心 (CEI) 中心主任感慨道。
在AlphaFold的幫助下,樸茨茅斯大學酶創新中心正在設計一種酶,來加速降解一次性塑料。
科羅拉多大學博爾德分校的一個團隊在使用AlphaFold來預測研究抗生素耐藥性的前景。
更多的可能性還在不斷被發掘。
業內不只有AlphaFold
除了AlphaFold和RoseTTAFold兩大工具以外,商業上也有與蛋白質預測相關的消息。
成立于2011年,專注于在大腸桿菌中制造蛋白質的公司Absci在周四上市。
今年1月,Absci收購了Denovium,一家使用深度學習人工智能對蛋白質進行分類和預測的公司,目前已經接受了1億種蛋白質的訓練。
Absci在6月還收購了Totient,一家分析免疫系統對某些疾病的反應的生物技術公司。
這樣一來,Absci就具備了蛋白質制造、評估和篩選能力,完善了從蛋白質制造和測試到人工智能支持的藥物開發。
隨著人工智能的不斷發展,人工智能有可能徹底改變21世紀的科學工作方式。
人工智能對蛋白質大規模的準確結構預測,將成為推動生物醫學界發展的重要工具,從蛋白質的結構解決新的科學問題。
熱切期待AlphaFold繼續幫助科學界解鎖下一步的發現。