哈佛、哥大開源1600萬組蛋白質序列,解決AlphaFold 2訓練數據私有難題!
蛋白質是生命的主力軍,了解它們的序列和結構,是設計新酶、開發救命藥物等生物學和醫學挑戰的關鍵。
DeepMind的AlphaFold 2,能夠以前所未有的準確性預測蛋白質結構。
然而,由于缺乏開放的訓練數據,這一領域的進展被嚴重阻礙。
但來自哈佛大學、哈佛醫學院、哥倫比亞大學、紐約大學和Flatiron Institute的研究者,引入了一個開源數據庫。
這個名為OpenProteinSet的開源數據庫,可以通過大規模提供蛋白質比對數據,來大大改善這種狀況。
它提供的數據集,和用于訓練AlphaFold 2的數據集質量相同。
因為AlphaFold 2,MSA的實用性爆炸性增長
蛋白質的功能,就編碼在氨基酸序列中。
在進化過程中,這些序列會積累一些微小的變化,而蛋白質的整體結構和功能卻一直保持不變。
多序列對齊(MSA)是一組和進化相關的蛋白質序列,通過插入間隙進行對齊,使匹配的氨基酸最終出現在同一列中。
通過分析這些MSA中的模式,可以深入了解蛋白質的結構和功能。
MSA的每一行,都是一個蛋白質序列。蛋白質是由20個氨基酸(或「殘基」)組成的一維字符串,每個氨基酸或「殘基」由一個字母表示。
目標或「查詢」 蛋白質在MSA的第一行中給出。后續行是根據與查詢序列的相似性,從大型序列數據庫中檢索到的進化相關(「同源」) 蛋白質。
為了改進比對、適應長度隨時間變化的同源序列,MSA比對軟件可以在同源序列中插入「缺口」(此處用破折號表示)或刪除殘基。
MSA中同源序列的數量(「深度」)及其多樣性,都有助于MSA的實用性。
 MSA引物
MSA引物
長期以來,MSA對蛋白質研究都至關重要,不過在2021年,因為AlphaFold 2的出現,MSA的實用性呈現了爆炸性增長。
通過MSA,AlphaFold 2能夠以近乎實驗級的準確性預測蛋白質結構。
然而有一個問題:雖然AlphaFold 2是開源的,但它的訓練數據仍然是私有的。
這樣做的計算成本很高。根據目標序列長度和正在搜索的序列數據庫的大小生成一個具有高靈敏度的MSA,可能需要幾個小時。
這樣,蛋白質機器學習和生物信息學的前沿研究除了少數大型研究團隊外,其他所有人都無法訪問。
1600萬個MSA全部開源
因此,團隊提出了OpenProteinSet,這是一個在AlphaFold 2及其以上規模訓練生物信息學的模型。
它包含了AlphaFold 2未發布的訓練集,包括所有唯一的蛋白質數據庫(PDB)鏈的MSAs和結構模板。
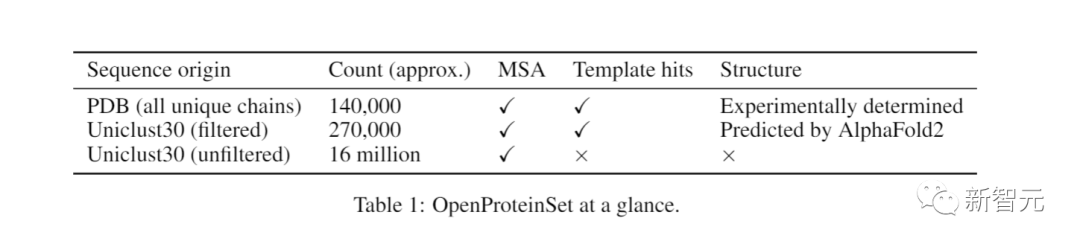
現在,OpenProteinSet提供了1600萬個MSA和相關數據,并且全部開源。
PDB是實驗確定的蛋白質結構的權威數據庫,而OpenProteinSet包括PDB中所有140,000種蛋白質的MSA。
它甚至還包括來自UniProt知識庫的序列,該序列按相似性聚類。
對于PDB蛋白質,OpenProteinSet能夠提供來自多個序列數據庫的原始MSA。
通過搜索PDB,它還能找到結構相似的蛋白質。
AlphaFold 2預測的結構,包括270,000個不同的UniProt集群。
使用開源數據集重新創建AlphaFold 2
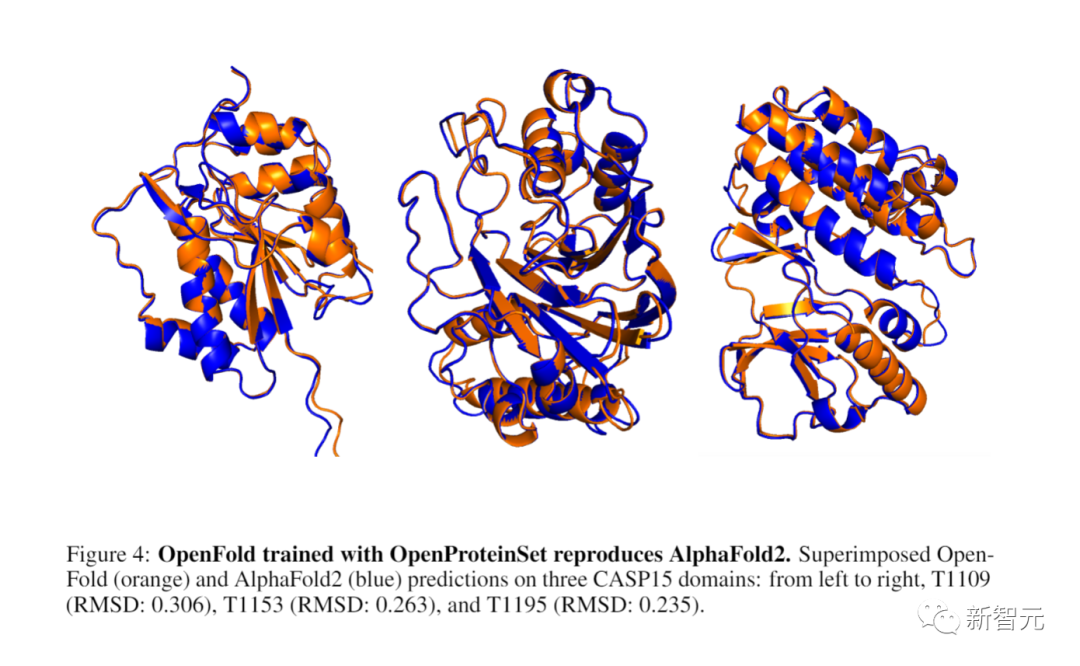
開發者還會使用OpenProteinSet來訓練OpenFold,這是AlphaFold 2的一個開放版本。
他們發現,OpenFold的性能與DeepMind的原始數據相當,證明了這種開放數據的充分性。
團隊表示,「通過OpenProteinSet,我們大大提高了分子機器學習社區可用的預計算MSA的數量和質量,」
該數據集可直接應用于結構生物學的各種任務。
實驗方法
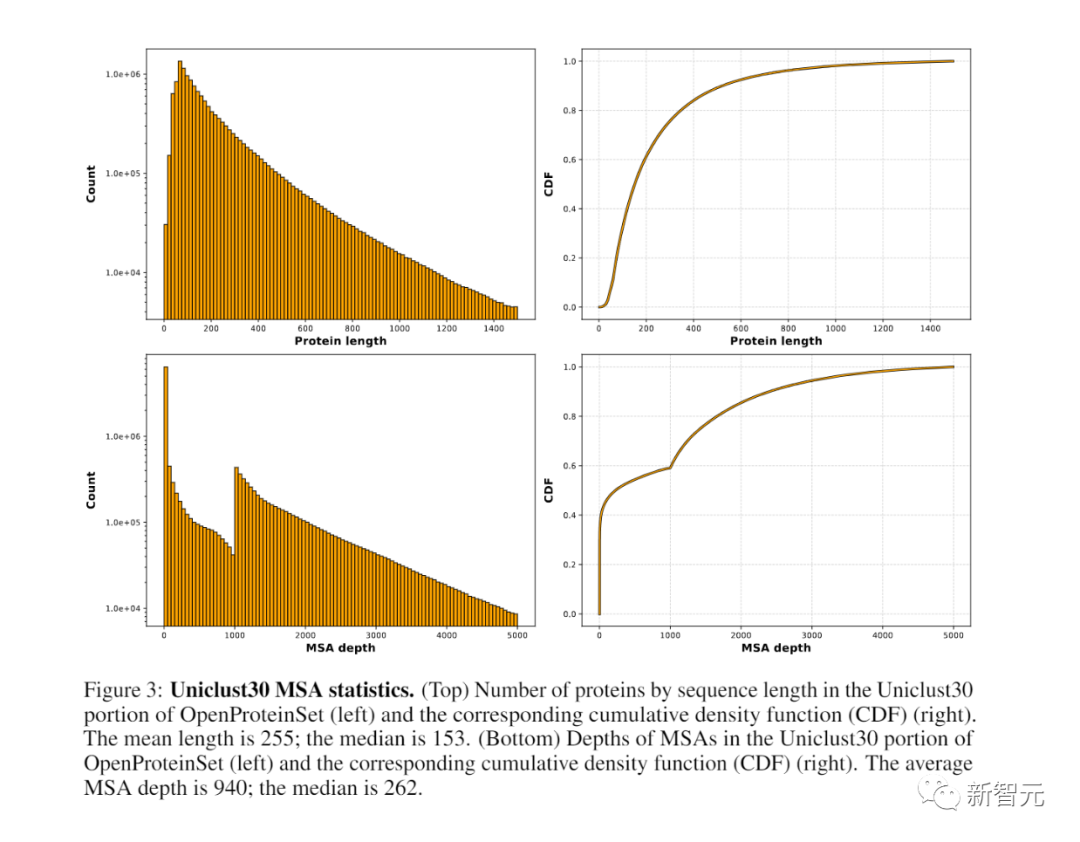
OpenProteinSet由超過1600萬個獨特的MSAs組成,這些MSAs是根據AIphaFold2論文中的程序生成的。
這一計數包括截至2022年4月PDB中所有14萬個唯一鏈的MSAs,以及針對同一數據庫為Uniclust30中的每個序列集群計算的1,600萬個MSAs。
從后一組中,研究者確定了270,000個最大多樣性代表性集群,比如可以適用于AphaFold2訓練過程中的自我蒸餾集。
對于每個PDB鏈,研究者使用了不同的對齊工具和序列數據庫計算三個MSAs。
使用OpenFold中的腳本,可以從公開可用的PDBmmCIF文件中,檢索相應的結構。
與用于生成AIphaFold2訓練集的過程一樣,研究者更改了MSA生成工具的一些默認選項。
隨后,產生了大約1600萬個MSAs,每個集群一個。
為了創建一個不同的、深度的MSAs子集,研究者通過迭代去除代表性鏈出現在其他MSAs中最多的MSAs。
這樣重復,直到每個代表鏈只出現在它自己的MSA中。
為了與對應的(未發布的)AlphaFold 2集進行奇偶性檢驗,研究者進一步刪除了代表序列大于1024個殘基或小于200個殘基的簇。
最后,他們剔除了相應MSAs少于200個序列的簇,只剩下270,262個MSAs。
總的來說,OpenProteinSet中的MSAs代表了超過400萬小時的計算。
OpenProteinSet大大提高了分子機器學習社區可用的預計算MSAs的數量和質量,它可以直接應用于結構生物學中的各種任務。
隨著模型對數據的需求越來越大,像OpenProteimnSet這樣的數據庫既可以作為多模態語言模型的生物知識寶庫,也可以作為多模態訓練本身的實證研究工具。
總之,OpenProteinSet將進一步推動生物信息學、蛋白質機器學習等領域的研究。