幾何機器學習:如何在基礎科學領域成為現實?
本文轉載自公眾號“讀芯術”(ID:AI_Discovery)
2020年,在幾何和圖形機器學習論文中表現突出的,當屬生物化學、藥物設計和結構生物學。這可能是第一次,我們終于發現這些機器學習方法對基礎科學的影響。本文中,我將重點介紹三篇論文,這三篇論文是過去一年內我感觸最深的論文(筆者是其中一篇論文的共同作者)。
幾何機器學習方法曾被刊登在《細胞》和《自然方法學》雜志2020年2月刊的封面上。
第一篇論文:
J. M. Stokes et al., A deep learning approach to antibiotic discovery (2020) Cell 180(4):688–702.
關于什么?基于圖形神經網絡研發抗菌藥物的深度學習操作流程。
如何操作?經訓練的圖神經網絡用于預測大腸桿菌在多于2000個已知抗菌活性分子(包括批準抗生素、動植物提取物)數據集上的生長抑制。這種預測只是基于分子圖,并不依賴于任何其他輔助信息,如藥物作用機制等。
訓練模型被送到藥物再利用中心,經調查研究,模型中含有約6000種藥物分子,前100種分子被選作試驗測試對象。令人吃驚的是,一種實驗抗糖尿藥物halicin(海利霉素)具備有效的抗菌效果,能夠消滅實驗小鼠體內的多種抗藥病菌。
顯然,圖神經網絡具備良好普適性,因為halicin分子不同于傳統抗生素。但是在這篇論文中,還并不清楚這種預測能力是否可以歸結為預測一種抗菌作用的簡單模式(細胞膜去極化)。
此外,研究人員還對ZINC15數據庫中超過1億個分子結構進行實驗篩選,ZINC15數據庫是專門為虛擬篩選而準備的商業可用化合物數據庫,通常為藥物設計者所用。在挑選的化合物中,物理試驗鑒定出8種具有抗菌活性,其中2種對多種病原體均有較強的活性。
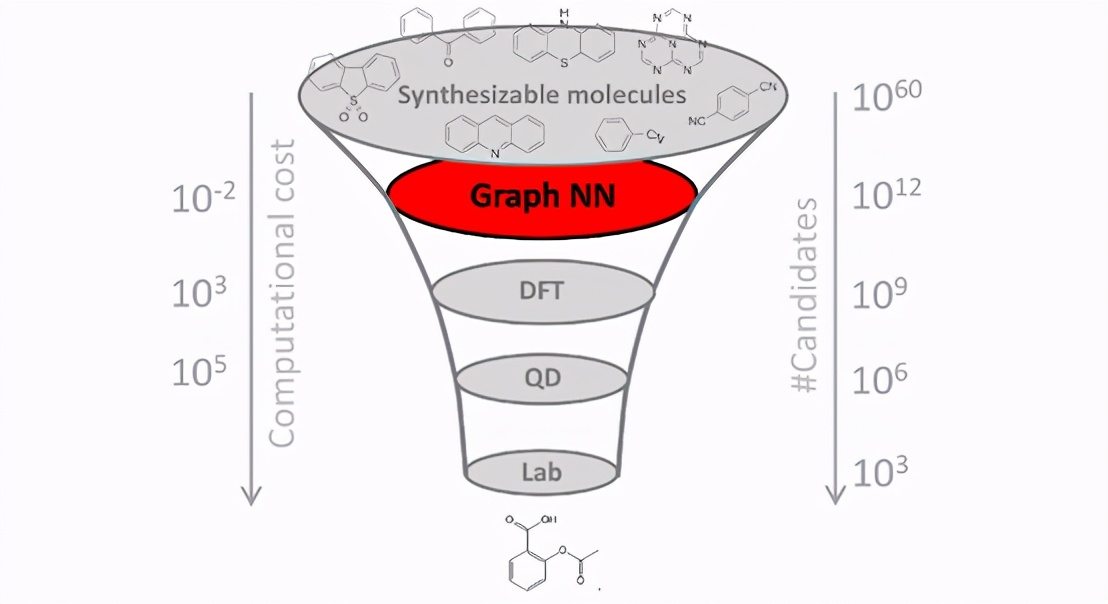
研發藥物的一大挑戰是,搜索空間很大,但是只有少數分子能夠在實驗室中測試。應用于分子圖的圖神經網絡可用來預測分子屬性,從而對所選藥物進行虛擬篩查。
為何重要?巨大的搜索空間是研發藥物的一大挑戰,據估計,其中至少包含1060個分子。只有很少一部分分子能夠在實驗室中測試,挑選較有可能性的分子至關重要。通過計算方法完成挑選的過程稱為“虛擬篩選”。
過去,機器學習方法經常用于分子的虛擬篩選,更廣泛來說,協助不同階段的藥物研發,這是第一次在完全沒有任何人類預假設的情況下,從零開始識別一種全新抗生素的過程。
大多數經由電腦模擬的、基于機器學習的藥物研發論文結果都僅由計算機預測,但是斯托克斯等人的論文與之不同,他們的論文不僅鑒別有潛力的藥物分子,而且在實驗動物上廣泛驗證它們在活體內的活性。
雖然在原則上這種方法只可以用于尋找治療癌癥等疾病的方法,但是對于抗生素的關注非常及時:濫用抗生素導致抗藥微生物形成,成為威脅全人類健康的夢魘,而且可能出現高傳染性的細菌感染,現有藥物無法治療,這個現象肯定會出現,只是時間問題而已。
更多相關內容:《量子雜志》熱搜文章和吉姆·柯林斯(Jim·Collins)2020年TED演講視頻(柯林斯實驗室是本年TED“無畏項目”之一,我們的CETI項目也屬于“無畏項目”)。
第二篇論文:
Jumper et al., High accuracy protein structure predictionusing deep learning (2020) a.k.a. AlphaFold 2.0 (尚未提供全文)
關于什么?根據氨基酸序列預測蛋白質的3D結構,這是生物信息學領域眾所周知的一個難題。
如何操作?AlphaFold 2.0是一個“基于注意力的神經網絡”(類似于變壓器結構),對蛋白質數據庫中17萬種蛋白質結構和未知結構蛋白質序列進行端對端訓練。但是DeepMind尚未公布算法細節,我們只能假設它是如何工作的。
在這篇文章中,蛋白質被建模為空間聯系圖,神經網絡“解釋該圖的結構,同時對正在構建的隱圖進行推理”。這聽起來很像具備潛在圖學習功能的圖神經網絡,只不過其中可能包含更多附加細節和細微差異,因為這種方法也使用進化序列信息,所以我將其歸類為“幾何機器學習”。
據報道,訓練的計算復雜度很高(相當于數年的GPU時間),而對結構的預測不過是“幾天的事”。
魔蛇玩具形象地展現了蛋白質折疊,在蛋白質折疊過程中氨基酸的一維序列折疊成復雜的3D形狀,賦予蛋白質功能
為何重要?蛋白質可以說是最重要的生物分子,經常被稱為“生命分子”,我們還未見過任何不以蛋白質為基礎的生命形式。蛋白質在DNA內編碼,在體內具備各種功能,包括抵抗病原體(抗生素)、形成皮膚結構(膠原蛋白)、輸送氧氣到細胞(血紅蛋白)、催化化學反應(酶)及信號傳遞(許多激素是蛋白質)。
從化學角度來講,蛋白質是生物聚合物或者由氨基酸組成的鏈,在靜電作用下折疊成復雜的3D結構。正是這種結構賦予蛋白質功能,而且這種結構對理解蛋白質是如何工作以及做什么是非常必要的。蛋白質一般是藥物治療的靶點(藥物是設計成與靶點相結合的小分子),所以制藥業極為關注該方面研究。
現代技術可以對蛋白質進行排列(即形成氨基酸串),而且成本較低、技術可靠,不過獲取3D結構主要還是依賴于傳統的結晶技術,盡管結晶技術不穩定、耗時長、成本高。目前,已知序列的蛋白質大約有2億種,已知結構的蛋白質至少有20萬種蛋白質。
一直以來,人們認為氨基酸序列中包含了足夠多預測蛋白質結構的信息,但是現在這個觀點站不住腳了。蛋白質結構預測關鍵技術分析大賽(CASP)是類似于ImageNet的競賽,自1994年開始舉辦,參賽者需要預測未知蛋白質的3D結構,這個大賽已成為生物信息實驗室和制藥公司的經典測試平臺。
2018年,DeepMind的新技術AlphaFold在CASP大賽中脫穎而出,獲得比賽勝利,震驚研究界。2020版AlphaFold 2.0效果更好,均方根誤差僅1.6埃,按照結構生物學標準可以說是非常精確,遠遠超過其他競爭對手。這是蛋白質科學領域里的“ImageNet時刻”。
盡管在關鍵問題上取得了驚人的進展,但是媒體大肆炒作、用詞隨意,歪曲了AlphaFold的功能。特別是在藥物設計應用上,結合部位通常需要達到亞埃精確度,但是這項技術尚未實現該功能。
更多相關內容:每個人都熱切期待解釋該算法的論文發表。萊克斯·弗里德曼(Lex Fridman)在YouTube視頻中進行了很好的概括,穆罕默德·艾爾庫雷希(Mohammed AlQuraishi)在博客中介紹了AlphaFold在2018年的影響。
第三篇論文:
P. Gainza et al., Deciphering interactionfingerprints from protein molecular surfaces using geometric deep learning (2020) Nature Methods 17(2):184–192.
關于什么?一個名為MaSIF的幾何深度學習方法從蛋白質的3D結構預測蛋白質之間的相互作用。
如何操作?MaSIF將蛋白質模擬為一個離散成網格的分子界面,研究人員認為此種方式在處理相互作用時是有利的,因為它可以提取出內部的折疊結構。這個架構是基于MoNet發明的,MoNet是我的博士研究生費德里科·蒙奇發明的一個網狀卷積神經網絡,基于預先計算的小地測片中的化學和幾何特點。
該網絡使用蛋白質數據庫中的幾千個共晶蛋白質3D結構來進行訓練,從而解決界面預測、配基分類和對接等各種問題,展現現代化的性能。MaSIF與其他方法最大的差異是,它不依賴于蛋白質的進化史。這在蛋白質全新設計中至關重要,嘗試“從頭”創造前所未有的全新蛋白質。
作為本篇論文的共同作者,我要強調的是預算分子界面和本地補丁的重要性,而且手工制作特性的依賴性是MaSIF的主要缺點之一。
在這一年里,我們徹底改造了結構,直接操作原子點云來輸入,飛速計算分子界面(表現為點云),學習幾何和化學特征,端到端可辨,運行速度快了幾個數量級(后者是通過使用快速幾何計算庫KeOps實現的,是我的博士后珍·菲迪(Jean Feydy)發明的)。
雖然《自然方法》論文主要關注計算方法,但是隨后EPFL的合作者獲得了MaSIF設計的幾種新型蛋白質結合劑的晶體結構,其與所計算結構高度吻合。
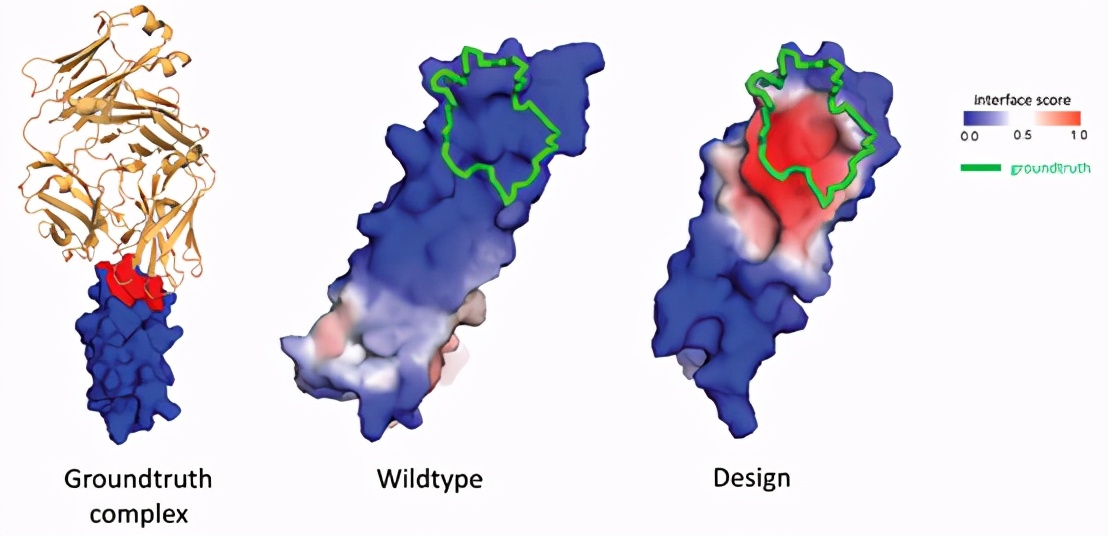
使用MaSIF預測蛋白質的結合位置。如圖所示設計蛋白質(右)經過修飾,以改善與自然產生的“野生型”(中)靶點相結合。即使結合部位結構偏平,MaSIF也可以準確探測其位置。
為何重要?蛋白質與其他生物分子之間的相互作用是大多數生物活動中蛋白質發揮功能的基礎。更好地理解蛋白質的作用原理對基礎生物學和藥物研發都非常重要,許多疾病與蛋白質間相互作用(PPI)有關,這種相互作用是理想的藥物靶點。然而,這種相互作用通常包含“不可藥物治療”的扁平界面,因為他們與傳統的小藥分子靶向的口袋型結構大不相同。
MaSIF能夠成功識別靶點的結合劑,是理性蛋白質設計的理想工具,開啟了生物藥物研究的各種應用,比如免疫抑制檢查站癌癥治療,這種療法以負責程序性細胞死亡的PD-1/PD-L1蛋白質復合體為靶體。